
中华医学会
地址: 中国北京东四西大街42号
邮编: 100710
先天性功能性孤立肾胎儿的遗传学病因:422例分析
本文引用格式:陈阳, 丁红珂, 卢建, 等. 先天性功能性孤立肾胎儿的遗传学病因:422例分析[J]. 中华围产医学杂志, 2025, 28(3): 185-193. DOI: 10.3760/cma.j.cn113903-20240307-00185.
摘要
目的 探讨先天性功能性孤立肾(congenital solitary functioning kidney, CSFK)的遗传学病因。
方法 本研究为回顾性研究,研究对象为2015年1月至2023年2月在广东省妇幼保健院因产前超声提示CSFK而接受介入性产前遗传学诊断的422例胎儿。分析CSFK胎儿各亚型(单侧肾缺如和单侧多囊性肾发育不良)、各组(孤立性和非孤立性CSFK)的G显带核型分析、染色体微阵列分析(chromosomal microarray analysis, CMA)和全外显子组测序(whole exome sequencing, WES)检测结果。采用χ2检验(或Fisher精确概率法)或秩和检验进行统计学分析。
结果 (1)422例胎儿母亲行介入性产前诊断时年龄为29(18~43)岁,孕周为25(17~34)周。234例接受了核型分析和CMA,63例接受了CMA和WES,7例同时接受了上述3种检测。故核型分析、CMA和WES分别完成了257、406和70例。(2)G显带核型分析的染色体异常检出率为1.6%(4/257)。这4例同时进行CMA检测,1例染色体嵌合胎儿样本未检出异常,3例染色体结构重排胎儿的CMA结果与G显带核型分析结果基本一致。(3)CMA的CNV检出率为8.9%(36/406),其中22例为致病性/可能致病性CNV。22例常见的CNV位点为17q12(5例)、22q11.21(5例)和16p11.2(2例);15例(68.2%)为微缺失综合征。这22例中的19例胎儿母亲选择引产终止妊娠,2例继续妊娠,1例失访。(4)共有241例同时进行了G显带核型分析和CMA检测(包括7例同时进行3种检测者),G显带核型分析和CMA的阳性率分别为1.7%(4/241)和5.8%(14/241)。与G显带核型分析相比,CMA可获得4.1%(10/241)的额外诊断率。(5)70例进行了家系WES检测,26例(37.1%)结果异常,其中致病/可能致病性12例,临床意义未明14例,WES阳性率为17.1%(12/70)。6例检出的基因变异(主要为PKD1和HNF1B基因变异)与胎儿CSFK表型有关,其中5例为常染色体显性遗传,1例为常染色体隐性遗传。在12例WES阳性病例中,杂合变异和复合杂合变异各6例;8例胎儿母亲选择继续妊娠,2例终止妊娠,2例失访。(6)422例CSFK胎儿中,35例(8.3%)检出遗传学异常。CSFK胎儿仅合并泌尿系统异常的遗传学异常阳性率最高,为15.1%(8/53),而CSFK胎儿合并泌尿系统以外的其他系统异常的阳性率为12.3%(7/57),孤立性CSFK胎儿的阳性率为6.4%(20/312)。3组比较,差异有统计学意义(χ2=5.95,P=0.048)。但组间多重比较时,差异无统计学意义。
结论 17q12微缺失综合征、22q11.2微缺失综合征、PKD1基因变异和HNF1B基因变异是CSFK胎儿的主要遗传学病因。
【关键词】 孤立肾;产前诊断;染色体畸变;回顾性研究
先天性功能性孤立肾(congenital solitary functioning kidney, CSFK)是指胎儿出生时1个肾脏在解剖或功能上的缺失,包括胚胎时期肾脏形成完全失败或发育异常导致功能缺失。CSFK可分为单侧肾缺如(unilateral renal agenesis, URA)和单侧多囊性肾发育不良(multicystic dysplastic kidney, MCDK)[1],是先天性泌尿系统畸形(congenital anomalies of kidney and urinary tract, CAKUT)的常见表型。CSFK是常见的出生缺陷之一,新生儿患病率为1/(1 000~2 000)[2-3]。胎儿CSFK主要在胎龄18~22周的超声检查中检出[4]。由于CSFK不影响胎儿生长发育、泌尿系统功能和羊水量,且肾上腺易误认为肾脏,故胎儿期CSFK易误诊。产前超声筛查CSFK的准确率仅为62%[6]。与双侧肾脏缺如或双侧MCDK胎儿的严重不良预后不同,既往研究表明CSFK儿童出现肾损伤的概率较小[7-8],因而产前超声提示胎儿存在CSFK时,多数孕妇仍选择继续妊娠[6,9]。但CSFK胎儿遗传学异常的概率有一定程度升高,故不良预后的发生率也可能升高[10]。已有研究探讨了染色体微阵列分析(chromosomal microarray analysis, CMA)和全外显子组测序(whole-exome sequencing, WES)在CSFK和CAKUT的应用[9,11],但对于CSFK的遗传学病因仍未得到明确结论。本研究拟回顾性分析在本单位行产前诊断的CSFK胎儿的遗传学检测结果和临床结局,探讨CSFK的遗传学病因。
资料与方法
一、研究对象
1.研究对象:本研究为回顾性研究。研究对象为2015年1月至2023年2月因产前超声提示CSFK,于广东省妇幼保健院医学遗传中心接受介入性产前诊断的胎儿424例。排除资料不全者2例后,其余422例胎儿纳入本研究。根据CSFK超声各亚型分类标准[4],将CSFK胎儿分为URA组(168例)和单侧MCDK组(254例);再根据是否合并其他异常,分为孤立性CSFK组(312例)和非孤立性CSFK组(110例),后者进一步分为仅合并泌尿系统异常亚组(53例)和合并非泌尿系统异常亚组(57例)。本研究经广东省妇幼保健院医学伦理委员会审查批准(202401048号),豁免知情同意。所有胎儿的母亲均接受检测前遗传咨询,同意进行介入性产前诊断。
2. CSFK的分型:参考文献[4]的标准。URA为一侧肾窝处未见肾脏回声,膀胱、羊水正常;单肾动脉;对侧肾脏代偿性肥大。单侧MCDK为一侧肾实质回声增强,被大小不一的非交通性囊肿取代;缺乏可识别的肾窦或肾实质组织。
二、研究方法
1.数据获取:通过本院病历系统检索2015年1月至2023年2月超声诊断为CSFK胎儿的病例,并从系统中获取这些病例的临床资料。(1)一般资料:包括孕妇年龄、孕周、孕产史、临床诊断、随访资料等。(2)检查资料:包括超声检查、G显带核型分析、CMA和WES检测结果等。这些检查均由本单位完成。
CMA采用美国Affymetric公司CytoScan 750K芯片平台进行检测。芯片扫描后数据导入染色体分析软件(chromosome analysis suite, ChAS)。WES数据获取:采用xGen Exome Research Panel v1.0(美国IDT公司)用于靶向基因组富集。在Illumina NovaSeq6000测序平台上进行测序,产生150 bp的末端配对序列。
2.数据分析:数据分析使用芯片平台配套的ChAS软件。结果参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南,将拷贝数变异(copy number variation, CNV)分为5个等级(致病性、可能致病性、意义未明、可能良性和良性)[12]。
使用BWA将序列映射到hg19/GRCh37上。使用GATK的HaplotypeCaller对变异和索引进行调用。VEP注释了功能结果,然后是AnnoVar注释。将每个变异与gnomAD、1000 Genomes、ESP6500和ExAC进行种群频率比较。用VarCards评估对蛋白质功能的影响。变异分类遵循2015 ACMG/美国分子病理学会(Association for Molecular Pathology, AMP)指南。
三、统计学分析
应用SPSS 22.0进行统计学分析。计数资料采用例数和百分数表示,采用χ2检验(或Fisher精确概率法)进行组间比较,采用Bonferroni方法进行多重比较。非正态分布的(样本含量>50,采用Kolmogorov Smirnov检验判断)计量资料采用M(P25~P75)表示,采用秩和检验进行组间比较。P<0.05(多重比较时为0.05/比较次数)为差异有统计学意义。
结果
一、一般资料
422例CSFK孕妇行介入性产前诊断时年龄为29(18~43)岁,孕周为25(17~34)周。检测标本包括羊水268例(63.5%)和154例(36.5%)。422例中,102例仅行CMA,16例仅行核型分析;234例接受了核型分析和CMA,63例接受了CMA和WES;7例同时接受了上述3种检测。故核型分析、CMA和WES分别完成了257、406和70例。见表1。
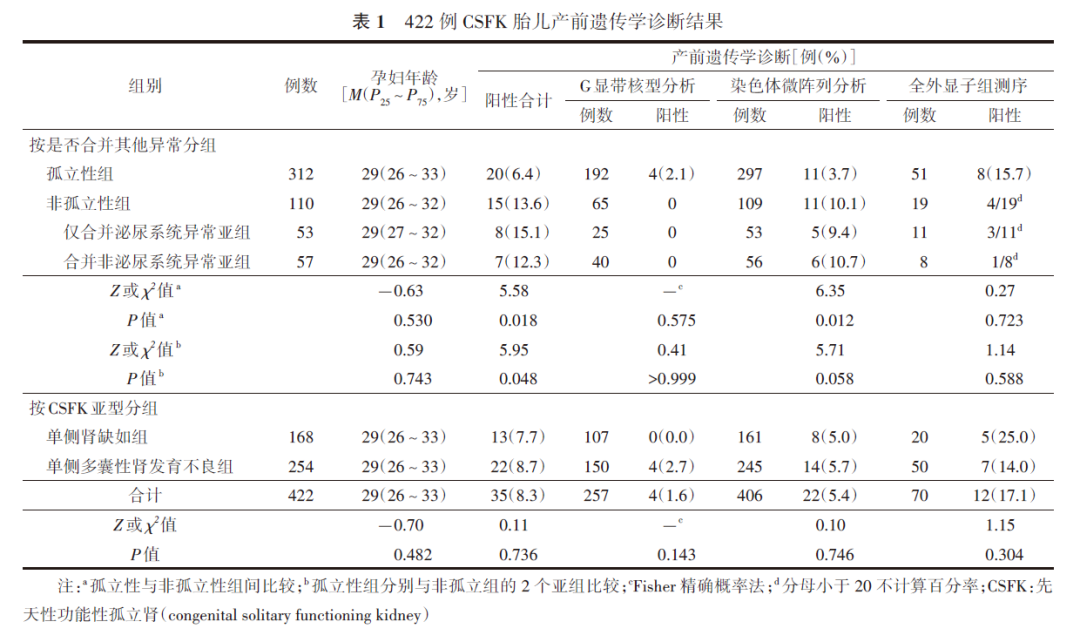
二、CSFK胎儿产前遗传学诊断结果
1.G显带核型分析结果:G显带核型分析的染色体异常检出率为1.6%(4/257),包括3例染色体结构重排和1例嵌合型额外小标记染色体。追踪该4例胎儿父母的染色体发现,其中2例父母未行此检测,另2例父母染色体核型均正常,胎儿染色体异常为新发变异。这4例胎儿样本均同时进行了CMA检测,结果1例染色体嵌合胎儿样本未检出异常,3例染色体结构重排胎儿的CMA结果与G显带核型分析结果基本一致(图1)。

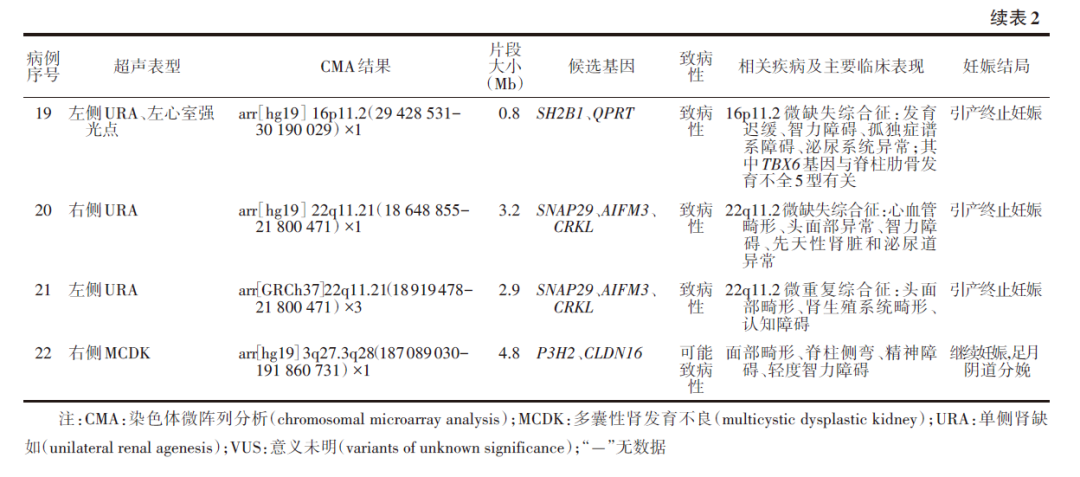
2.CMA检测结果:406例胎儿进行了CMA检测,检出36例(8.9%)CNV,其中22例为致病性/可能致病性CNV。22个CNV片段的大小为2.8(0.3~58.1) Mb,其中≥5 Mb者5例,片段大小为22.5(8.7~58.1) Mb;<5 Mb的微小CNV片段17例,片段大小为1.6(0.3~4.8) Mb。22例常见的CNV位点为17q12(5例)、22q11.21(5例)和16p11.2(2例);15例(68.2%)为微缺失综合征,包括17q12微缺失综合征、22q11.2微缺失综合征、16p13.3重复综合征和16p11.2微缺失综合征等。19例胎儿母亲选择引产终止妊娠,2例继续妊娠,1例失访。见表2。

本研究中,有241例同时进行了G显带核型分析和CMA检测(包括7例同时进行3种检测者),G显带核型分析阳性率为1.7%(4/241),CMA阳性率为5.8%(14/241)。相对于G显带核型分析,CMA可获得4.1%(10/241)的额外诊断率。
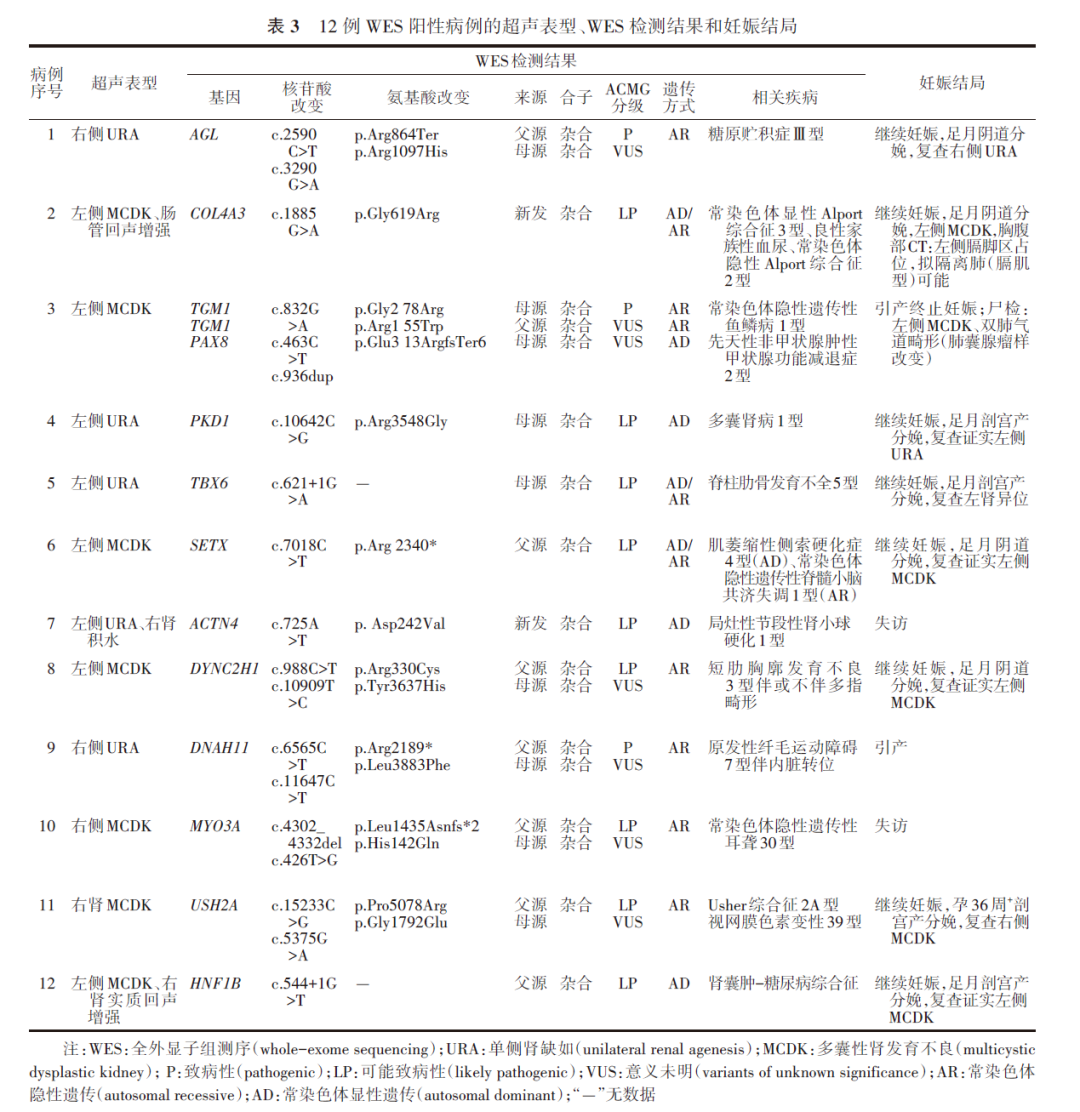
3.WES检测结果:70例进行了家系WES检测,26例结果异常,其中致病/可能致病性基因变异12例,意义未明变异14例,WES阳性率为17.1%(12/70)。在结果异常的病例中,可导致肾脏疾病的基因变异有PKD1、PKD2、HNF1B、HNF1、PAX2、DYNC2H1、GANAB、COL4A3、SIX5、GANAB、NEK8、ACTN4和FREM1。其中,有6例检出的基因变异与胎儿CSFK表型有关,变异基因分别为HNF1B(c.544+1G>T)、HNF1B(c.313G>A)、PKD1 (c.10642C>G)、PAX2(c.461C>T)、DYNC2H1 (c.988C>T)和GANAB(c.2833T>A);5例为常染色体显性遗传,1例为常染色体隐性遗传。
在12例WES阳性病例中(表3),杂合变异和复合杂合变异各6例;检出新发变异2例、遗传自父亲或母亲各2例,以及父母双亲6例;遗传方式包括常染色体隐性遗传6例和常染色体显性遗传3例,以及常染色体隐性/显性遗传3例;8例孕妇选择继续妊娠、2例终止妊娠、2例失访。
4.CSFK胎儿产前遗传学诊断结果及各亚型、分组的差异:422例CSFK胎儿中,35例(8.3%)检出遗传学异常。各亚型中,单侧MCDK的阳性率高于URA[分别为8.7%(22/254)和7.7%(13/168)],但差异无统计学意义(P>0.05)。各分组中,CSFK胎儿仅合并泌尿系统异常的遗传学异常阳性率最高,为15.1%(8/53),而CSFK胎儿合并泌尿系统以外的其他系统异常的阳性率为12.3%(7/57),孤立性CSFK胎儿的阳性率为6.4%(20/312)。3组比较,差异具有统计学意义(χ2=5.95,P=0.048)。见表1。对这3组采用Bonferroni方法进行多重比较,差异无统计学意义。
讨论
本研究对CSFK胎儿的遗传学病因进行了探讨。通过应用不同检测技术,本研究发现CSFK胎儿的预后与是否合并其他异常,以及遗传学病因密切相关。本研究中,G显带核型分析、CMA 检测和 WES 检测的阳性率分别为1.6%(4/257)、5.4%(22/406)和17.1%(12/70)。CSFK胎儿最常见的遗传学异常是17q12和22q11.2微缺失综合征,以及PKD1和HNF1B基因变异。本研究还发现,孤立性与非孤立性CSFK胎儿的遗传学异常发生率存在差异,合并泌尿系统外或其他泌尿系统异常的CSFK胎儿遗传物质发生变异的风险明显升高。
一、CSFK与G显带核型分析
在对CSFK胎儿遗传学病因的探究中,传统G显带核型分析技术的遗传学异常检出率较低。Fu等[13]和Cai等[14]分别对72例和108例MCDK胎儿进行研究,发现染色体核型异常率检出率分别为4.2%和3.7%。本研究对257例CSFK胎儿进行核型分析,仅发现3例染色体结构重排及1例嵌合体,阳性率为1.6%,与上述文献结果相近。本研究G显带核型分析发现1例嵌合型额外小标记染色体胎儿。文献报道70%的额外小标记染色体携带者表型正常[15],该病例经CMA检测未发现异常拷贝数变异,且出生后随访临床表型亦未见异常,由此推测该病例的额外小标记染色体更可能为异染色质区。从遗传学角度看,这种染色体结构特性可能与胎儿相对正常的表型存在关联。与合并其他超声异常胎儿的染色体核型分析技术检出率(7.3%)相比[16],CSFK胎儿的检出率更低。这主要归因于G显带核型分析技术本身的局限性导致该方法难以精准检测微缺失微重复的染色体异常,而这些类型恰是CSFK 胎儿常见的遗传学病因类型。基于此,对于产前超声提示CSFK的胎儿,CMA检测在遗传学病因诊断方面更具优势,可能可以作为替代传统核型分析技术的有效手段,以更精准地探测染色体的细微结构变化,从而为明确CSFK胎儿的遗传学病因提供更可靠的依据。
二、CSFK与CMA检测
CMA检测技术在CSFK胎儿遗传学病因探索中发挥关键作用。CMA能够有效检出染色体结构的微缺失/微重复异常,并明确异常片段内的致病基因。本研究对406例胎儿进行CMA检测,发现22例胎儿存在致病性/可能致病性CNV,阳性率为5.4%,低于既往文献报道[9,17]。分析其原因,一方面,本研究纳入病例的数量与亚型存在差异,本研究纳入的406例胎儿包含URA及单侧MCDK胎儿,而文献纳入者均为MCDK胎儿;另一方面,本研究未纳入双侧MCDK胎儿,而文献中双侧MCDK胎儿有30例且CMA异常率高达30%。
本研究发现的主要异常位点包括17q12和22q11.21(各5例),以及16p13.11和16p11.2(各2例),与Sanna等[18]的研究发现相近,印证了这些染色体片段异常是CAKUT疾病常见的遗传学病因所涉及的位点。从遗传学致病机制来看,17q12微缺失综合征的致病基因主要为HNF1B和LHX1基因[19-21],85%~90%的个体因这些基因异常而出现肾脏和泌尿道的结构或功能异常;约40%的患者出现青少年5型成熟期发病糖尿病(maturity-onset diabetes of the young type 5, MODY5);约50%的患者存在一定程度的神经发育或神经精神疾病(发育迟缓、智力障碍、孤独症谱系障碍、精神分裂症、焦虑症和双相情感障碍等)。本研究中的5例17q12微缺失综合征胎儿均表现为单侧MCDK,且3例合并对侧肾实质回声增强,这一超声特征可作为该综合征在产前诊断中的重要线索。染色体22q11.2微缺失是Di George综合征、腭-心-面综合征和圆锥动脉干-异常面容异常综合征的分子病理基础,主要临床表现为心血管畸形、头面部异常、智力障碍、先天性肾脏和泌尿系统异常[22]。该片段内相关致病基因(如SNAP29、AIFM3和CRKL)的变异与泌尿系统异常密切相关,其中CRKL单倍剂量不足是主要遗传致病因素[23]。本研究中有1例16p13.11微重复和1例16p13.11微缺失的单侧MCDK胎儿。16p13.11微重复因基因剂量变化与行为异常、发育迟缓、先天性心脏缺陷和骨骼异常等多种临床特征相关;16p13.11微缺失则与智力障碍和癫痫相关[24]。该2例胎儿的相同异常染色体区域包含的ABCC6基因在小鼠肾脏近端小管的基底膜中高表达并行使转运蛋白功能[25]。虽然其剂量敏感致病机制尚未明确,但提示该基因在CSFK胎儿肾脏发育异常的遗传学病因中可能发挥重要作用。
本研究中的2例16p11.2微缺失综合征胎儿表型均为URA。文献报道,16p11.2缺失的个体出现精神和发育障碍的频率较高(>90%)[26],该区域内SH2B1和QPRT基因缺失可能是导致肾脏异常的遗传学因素,QPRT缺失可通过WNT通路引发肾小管异常和继发性肾小球囊肿;SH2B1是作为接头蛋白与RET酪氨酸激酶活性增强有关,而RET与肾脏发育异常紧密相连,进一步证实了这些基因异常在CSFK胎儿遗传学病因的中的重要性[11,27]。
三、CSFK与WES检测
在探究CSFK胎儿遗传学病因中,单基因变异作为关键节点可显著干扰肾脏发育进程。遗传学已成功甄别出50余个与CAKUT病因紧密交织的基因[18,28],而WES技术逐渐在CSFK胎儿产前遗传学诊断领域崭露头角,成为提升遗传学病因检出率的有力工具。van der Ven等[29]使用WES在29个(13%,29/232)家庭中检测到与CAKUT有关的已知基因的致病性变异。Cai等[14]使用WES在13例MCDK胎儿中发现2例存在BBS1和BBS2基因变异。本研究显示,CSFK胎儿排除染色体变异外,其基因发生致病性/可能致病性变异的概率也较高(17.1%,12/70)。有6例检出的基因变异与胎儿肾脏异常表型有关,其致病基因分别为PDK1、DYNC2H1、GANAB、PAX2和HNF1B,均与肾脏发育有关,为CSFK已知致病基因。该6例胎儿的基因变异均来源于父母。针对父母泌尿系统情况进一步随访发现,有3例胎儿父母之一双侧肾脏均存在异常。因此,当CSFK胎儿存在基因变异时,除了代偿机制引起对侧肾脏增大外,还可能因遗传变异导致对侧肾脏异常,需严格监测胎儿对侧肾脏。
四、CSFK胎儿不同亚型、分组的遗传学异常差异
本研究在8.3%(35/422)的CSFK胎儿中检出遗传学异常。各亚型遗传学异常检出率差异并无统计学意义,然而,CMA检查结果显示,17q12微缺失综合征均出现在单侧MCDK胎儿中,16p11.2微缺失综合征均出现在URA胎儿中。这一结果提示单侧MCDK与URA可能存在不同的发病机制和遗传学异常,但还需更大样本研究进一步确认。在分组分析中,孤立性与非孤立性CSFK胎儿的遗传学异常发生率存在明显差异。非孤立性CSFK胎儿,尤其是仅合并泌尿系统异常组的遗传学异常发生率最高。这可能是多种因素共同作用的。一方面,合并其他异常可能意味着胎儿在发育过程中受到更多的干扰因素,增加了遗传物质发生变异的可能性。另一方面,不同的异常表现可能反映了不同的信号通路或机制的异常。例如,合并泌尿系统外异常可能涉及多个器官系统的发育调控基因异常,而仅合并泌尿系统异常可能与泌尿系统特定发育阶段的关键基因异常有关。为了更好地理解CSFK胎儿的遗传学基础和发病机制,未来的研究可以进一步探讨不同分组中具体的遗传学异常类型,以及这些异常与胎儿临床表现之间的关系;也可以结合功能研究和动物模型等手段,深入研究关键基因的作用机制,为临床诊断和治疗提供更有力的依据。
本研究存在一定局限性。一方面,本研究未综合分析胎儿父母CMA结果,对CNV来源及家系分析有一定局限性;另一方面,本研究未对继续妊娠的CSFK胎儿进行远期随访,所以遗传学未见异常的CSFK胎儿的远期预后仍需要进一步研究。
本研究发现,17q12微缺失综合征、22q11.2微缺失综合征、PKD1基因变异和HNF1B基因变异是CSFK胎儿的主要遗传学病因。CSFK胎儿的遗传学异常率较高,且孤立性CSFK、合并除泌尿系统外的其他系统异常和合并其他泌尿系统异常的遗传学变异风险依次升高。染色体变异或基因变异不仅与胎儿肾脏发育异常相关,还可能影响到其他系统,如神经智力发育等。建议对CSFK胎儿均行产前遗传学诊断。如条件允许,建议联合WES技术,可能有助于提高CSFK胎儿遗传学异常检出率。
利益冲突 所有作者声明不存在利益冲突



